Синдром Барттера является одним из самых редких генетических заболеваний, при котором идет разрушение почечных канальцев в процессе разрастания юкстагломерулярного аппарата с высоким уровнем секреции ренина и альдестерона.
Симптомы
Еще в антенатальном периоде СБ-отклонения обнаруживают себя повышенной концентрацией хлорида в амниотической жидкости.
Типичная клиника у новорожденных – пониженная масса тела, полиурия с риском обезвоживания, лихорадка и судороги.
При выявлении подобной симптоматики следует сразу же записаться на прием к врачу для проведения внепланового обследования ребенка.
Новорожденные с подозрением на СБ-изменения отличаются плохим аппетитом, сонливостью. Внешние отличия: выступающий лоб, оттопыренные ушные раковины, большие глаза.
В последующем главными нарушениями обмена веществ в организме остаются гиперкальциурия с нефрокальцинозом, гипокалиемия и остеопения.
СБ-классическое течение клинически проявляет себя в раннем детском возрасте эпизодами мышечной слабости и подергивания, диареи, лихорадки.
Ввиду выраженной полиурии у таких пациентов возникает риск появления гидронефроза, а хроническая гипокалиемия сопровождается образованием кисты.
Врачи, которые могут помочь

Виды
Сегодня существуют следующие виды заболевания синдрома Барттера:
1) Неонатальный;
2) Классический.
Причины
Данное заболевание очень часто передается по наследству с отличием аутосомно-рецессивного типа, но существуют и другие причины этого заболевания.
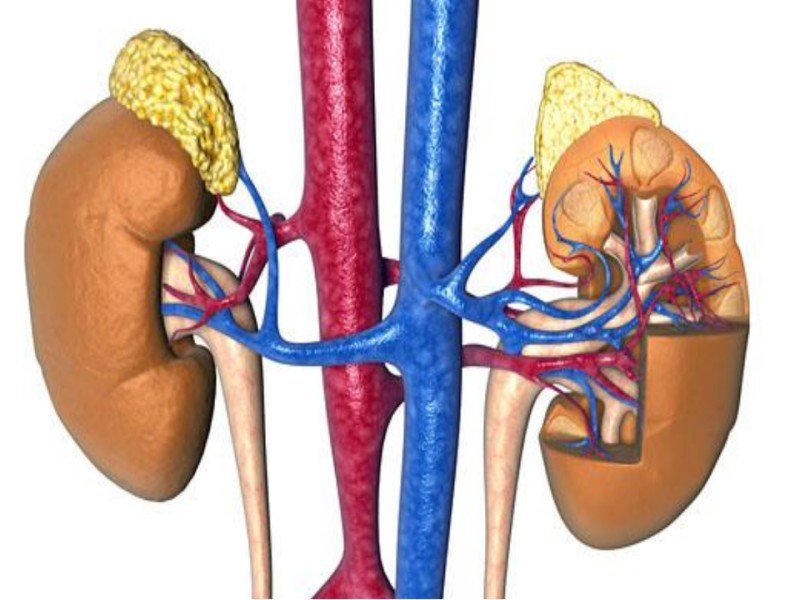
За этиологию патогенного заболевания синдрома Барттера отвечает мутация генов котранспортера почек, который находится в отделе Генлевской петли. Заболевание провоцирует расстройства, связанные с реабсорбцией натрия, понижения внутреннего объема сосудов и увеличения продуцирования таких веществ, как ренин и альдестерон.
Развитие классического-СБ происходит при кодировке дефектным геном почечного хлорного канала. В результате нарушается реабсорбция хлорида натрия.
Диагностика
Заподозрить заболевание возможно еще во внутриутробный период, когда прослеживается чрезмерное увеличение количества амниотической жидкости.
В дальнейшем СБ - диагностика заключается в тщательном сборе акушерского анамнеза, выявлении специфической симптоматики у новорожденных детей. Обращается внимание на их слабость, отсталость в росте, присутствие признаков дегидратации и характерных визуальных критериев болезни.
На первой неделе жизни лабораторные исследования обнаруживают гипокалиемию и метаболический алкалоз. При низком удельном весе мочи в ней фиксируется превышение Ca, Na, Cl, K и простагландинов.
Анализ крови показывает высокий уровень альдостерона и ренина. Вследствие продолжительной гиповолемии иногда удается идентифицировать гиперурикемию.
Изменения в почечной паренхиме и сосудистом русле фиксируются с помощью ультразвукового исследования и компьютерной томографии.
Дифференциальная диагностика проводится в отношении гипокалиемического алкалоза, развивающегося из-за чрезмерного употребления диуретиков, и кишечной потери калия и натрия.
По показаниям проводятся консультации педиатра, нефролога, акушер-гинеколога, генетика, уролога.
Лечение
Терапия неонатального этапа направлена на коррекцию метаболических показателей и устранение дегидратации с помощью инфузий изотонического раствора хлорида натрия и восполнения хлорида калия. Не раньше 4-5 недели жизни назначается индометацин.
Индометацин также используется для излечения классического-СБ. Рекомендуется восполнение потерь калия назначением КС1 в сочетании со верошпироном (спиронолактоном).
Рациональные терапевтические мероприятия в короткие сроки нормализуют клиническое состояние пациента. Однако для большинства больных они носят прижизненный характер.
В процессе медикаментозной терапии особое внимание уделяется снижению гастроинтестинальной, печеночной и почечной токсичности индометацина.
Профилактика
Для СБ-профилактики целесообразно проведение медико-генетических консультаций лиц, семейный анамнез которых отягощен недугом.